torch-sim¶
torch-sim is a tool for running (batched) molecular dynamics and structure optimisations in a GPU-accelerated manner.
You can use any model from graph-pes with torch-sim! Doing this is extremely straightforward, as this notebook hopes to demonstrate. One caveat is that torch-sim expects all models to return energies and stresses in units of eV and forces in units of eV/Å. If you have a model that was trained in a different unit system, you will need to wrap it in a graph_pes.models.UnitConverter object so that
it can be used in torch-sim.
To get started, you must first ensure you have the torch-sim-atomistic package installed:
[ ]:
# this notebook ran successfully with this torch-sim version.
# future versions may require changes as the package evolves.
!pip install graph-pes torch-sim-atomistic==0.2.0
[1]:
from pathlib import Path
import ase
import ase.io
import load_atoms
import torch
import torch_sim
from ase.build import bulk
from graph_pes import models
%config InlineBackend.figure_format = 'retina'
torch.manual_seed(42) # for reproducibility
DEVICE = torch.device("cuda" if torch.cuda.is_available() else "cpu")
print(f"Using device: {DEVICE}")
[1]:
Using device: cpu
NVT MD¶
torch-sim makes it easy to run NVT molecular dynamics using a Langevin thermostat.
To keep things simple, we use a simple LennardJones model to run NVT for a single structure. It’s worth repeating that this proceduce will be identical for any other model in graph-pes, be that a model you’ve trained from scratch yourself, a pre-trained foundation model or a foundation model you have fine-tuned on your own
data.
[2]:
# make a model: this could be any model from `graph-pes`
model = models.LennardJones(sigma=1.5, epsilon=1.0)
ts_model = model.torch_sim_model(device=DEVICE)
# create a bulk Cu structure using ASE
structure = bulk("Cu", "fcc", a=3.58, cubic=True).repeat(4)
# NVT at 300 K with a timestep of 0.002 fs
torch_sim.integrate(
system=structure,
model=ts_model,
n_steps=500,
timestep=0.002,
temperature=300,
integrator=torch_sim.nvt_langevin,
trajectory_reporter=torch_sim.TrajectoryReporter(
"Cu-traj.h5md",
state_frequency=10, # save every 10 steps
state_kwargs=dict(save_velocities=True, save_forces=True),
),
)
# post-process the trajectory to save as an ASE-compatible file
trajectory = torch_sim.TorchSimTrajectory("Cu-traj.h5md")
trajectory.write_ase_trajectory("Cu-traj.traj")
load_atoms.view(ase.io.read("Cu-traj.traj", index="-1"))
[2]:
Of course, a Lennard-Jones model is not particularly realistic for this case, but we can see that MD has generated a collection of Lennard-Jones clusters as expected.
torch-sim saves structure snapshots in a custom format. To extract ase.Atoms objects with the force and velocities properties, we can use the following helper functions:
[3]:
def get_atoms_with_labels(trajectory: torch_sim.TorchSimTrajectory, index: int):
atoms = trajectory.get_atoms(index)
for property in ["forces", "velocities"]:
if property not in trajectory.array_registry:
continue
atoms.arrays[property] = trajectory.get_array(
property, index, index + 1
)[0]
return atoms
def trajectory_to_ase_structures(file_name: str | Path) -> list[ase.Atoms]:
traj = torch_sim.TorchSimTrajectory(file_name)
atoms = []
i = 0
while True:
try:
atoms.append(get_atoms_with_labels(traj, i))
except IndexError:
break
i += 1
return atoms
structures = trajectory_to_ase_structures("Cu-traj.h5md")
structures[0].arrays["forces"].shape
[3]:
(256, 3)
NPT MD¶
torch-sim also supports NPT molecular dynamics using a Langevin barostat and thermostat. Below, we do just that.
Note that graph-pes models return stresses in the energy and length-scale units that they were originally trained on: typically this is eV and Å, but not always. Use a graph_pes.models.UnitConverter to convert the energies to eV and lengths to Å if necessary.
torch-sim expects the external pressure provided to be in units of eV/Å^3.
[4]:
amorphous_carbon_structures = load_atoms.load_dataset("C-GAP-17").filter_by(
lambda structure: len(structure) < 50,
config_type="bulk_amo",
)[:100]
amorphous_carbon_structures
[4]:
Dataset:
structures: '100'
atoms: 3,552
species:
C: 100.00%
properties:
per atom: (forces)
per structure: (config_type, detailed_ct, energy, split)
Here we load in a model trained on part of the C-GAP-17 dataset:
[5]:
from graph_pes.utils.analysis import parity_plot
carbon_model = models.load_model("c-gap-17-painn.pt")
parity_plot(
carbon_model,
amorphous_carbon_structures,
"forces",
lw=0,
s=3,
units="eV/Å",
)
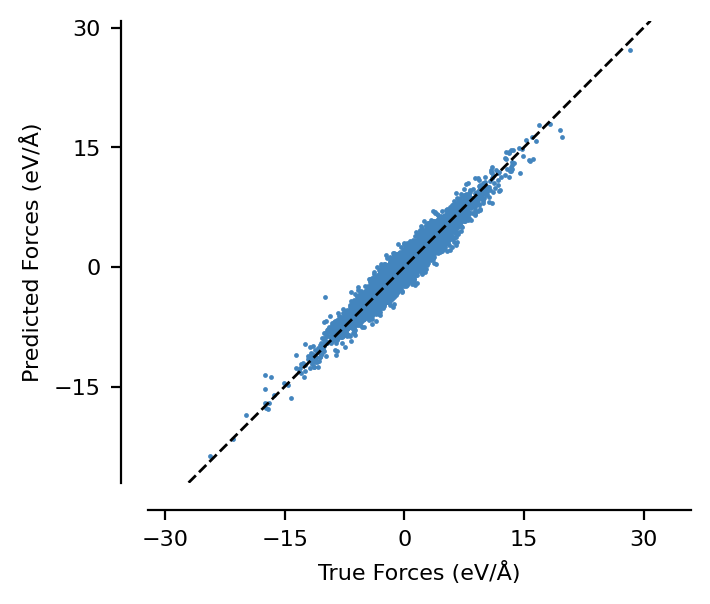
We can see that this model generates stress predictions as expected - graph-pes automatically calculates stresses for all models, allowing them to be used in NPT simulations.
[6]:
carbon_model.ase_calculator().get_stress(amorphous_carbon_structures[0])
[6]:
array([ 0.07337982, 0.06381519, 0.08241484, 0.00042613, -0.01667964,
0.00498147])
We start with a low-density amorphous carbon structure:
[7]:
starting_structure = amorphous_carbon_structures[0].copy()
load_atoms.view(starting_structure, show_bonds=True)
[7]:
[ ]:
import numpy as np
import matplotlib.pyplot as plt
# negative sign shows that we want to **compress** the system
desired_pressure = -1000 # in bar
bar_to_eV_per_A3 = 6.2415e-07
print(f"Starting cell:\n{starting_structure.cell.array.round(2)}\n")
# run NPT
final_state = torch_sim.integrate(
system=starting_structure,
model=carbon_model.torch_sim_model(device=DEVICE),
n_steps=1_000,
timestep=0.001,
temperature=300,
integrator=torch_sim.npt_langevin,
external_pressure=-desired_pressure * bar_to_eV_per_A3,
trajectory_reporter=torch_sim.TrajectoryReporter(
"test.h5md", state_frequency=10
),
b_tau=0.1,
)
trajectory = torch_sim.TorchSimTrajectory("test.h5md")
cells = trajectory.get_array("cell")
print(f"Final cell:\n{cells[-1].round(2)}")
Starting cell:
[[9.1 0. 0. ]
[0. 9.1 0. ]
[0. 0. 6.83]]
Final cell:
[[7.86 0. 0. ]
[0. 7.86 0. ]
[0. 0. 5.9 ]]
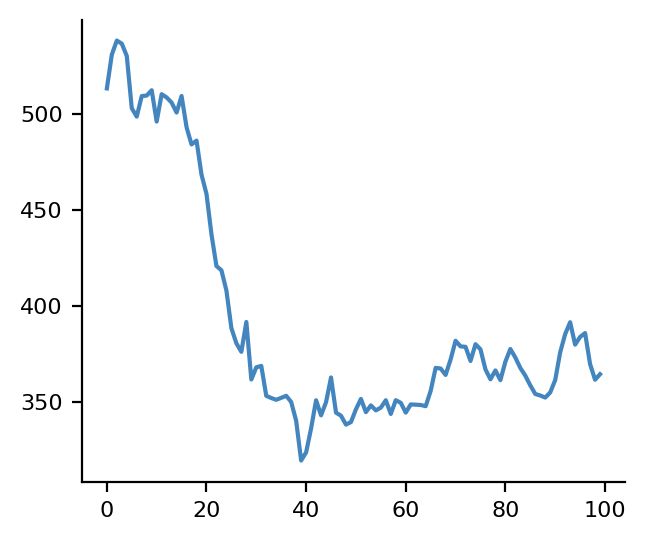
We can see that the NPT simulation has compressed the system as expected
[9]:
volumes = np.linalg.det(cells)
plt.plot(volumes)
[9]:
[<matplotlib.lines.Line2D at 0x307389a50>]
[10]:
load_atoms.view(final_state.to_atoms()[0], show_bonds=True)
[10]:
You can also express a (3, 3) shaped torch.Tensor as an anisotropic external pressure. See the torch-sim docs for more details.
Relaxation¶
Relaxation in torch-sim is provided via torch_sim.optimize(). We relax the final state of the NPT simulation:
[ ]:
relaxed_state = torch_sim.optimize(
system=final_state,
model=carbon_model.torch_sim_model(device=DEVICE),
optimizer=torch_sim.frechet_cell_fire,
)
load_atoms.view(relaxed_state.to_atoms()[0], show_bonds=True)
Batched simulations¶
The real power of torch-sim comes when you want to run many simulations at once.
Here’s a simple example of how to run NVE MD for a collection of structures simultaneously. Crucially, you can use this batching process with any of the above examples too!
[12]:
from graph_pes.interfaces import mace_off
# use the MACE-OFF (small) model to run NVE simulations
# since these structure have no unit cell, it is important to set
# compute_stress=False to avoid an error
mace_off_torch_sim = mace_off("small").torch_sim_model(
device=DEVICE, dtype=torch.float32, compute_stress=False
)
Using MACE-OFF23 MODEL for MACECalculator with /Users/john/.cache/mace/MACE-OFF23_small.model
[13]:
# load the first 100 structures from the QM7 dataset
structures = load_atoms.load_dataset("QM7")[:100]
# run batched NVE simulations for multiple structures simultaneously
# letting torch-sim handle batching and memory management for us
final_states = torch_sim.integrate(
system=list(structures),
model=mace_off_torch_sim,
n_steps=100,
timestep=0.001,
temperature=300,
integrator=torch_sim.nve,
trajectory_reporter=torch_sim.TrajectoryReporter(
# save each trajectory to a unique file
[f"QM7-traj-{i}.h5md" for i in range(len(structures))],
state_frequency=10,
),
)