Implement a model¶
FYI, you can open this notebook in Google Colab and follow along interactively 😊
Any and all GraphPESModel subclasses are fully compatible with graph-pes! You can:
train them using
graph-pes-trainrun MD in LAMMPS using
pair_style graph_pesload and analyse them using load_model
To demonstrate this, below we’ll implement a somewhat arbitrary model, and train it on the QM7 dataset.
Implementation¶
This model generates embeddings of neighbouring atoms as a function of their distance from the central atom and their atomic number.
These embeddings are summed over all neighbouring atoms to generate an embedding of the local environment, before being read out to predict the local energy:
[3]:
%%writefile custom_model.py
from __future__ import annotations
import torch
from graph_pes import AtomicGraph, GraphPESModel
from graph_pes.atomic_graph import (
PropertyKey,
index_over_neighbours,
neighbour_distances,
sum_over_neighbours,
)
from graph_pes.models.components.distances import Bessel, PolynomialEnvelope
from graph_pes.utils.nn import MLP, PerElementEmbedding
class CustomModel(GraphPESModel):
def __init__(
self,
cutoff: float,
channels: int,
radial_features: int,
):
super().__init__(
cutoff,
implemented_properties=["local_energies"],
)
# node embeddings
self.Z_embedding = PerElementEmbedding(channels)
# messages
self.radial_basis = Bessel(radial_features, cutoff)
self.envelope = PolynomialEnvelope(cutoff)
self.message = MLP(
layers=[
radial_features + channels,
2 * channels,
2 * channels,
channels,
],
activation="CELU",
bias=False,
)
# readout
self.readout = MLP(
layers=[channels, channels, 1],
activation="CELU",
)
def forward(self, graph: AtomicGraph) -> dict[PropertyKey, torch.Tensor]:
# compute the local energies
# assume that the input graph has N atoms, E edges
# and denote the number of radial features as R
# and the number of channels as C
# 1. embed node features
h = self.Z_embedding(graph.Z) # (N, C)
# 2. expand distances
r = neighbour_distances(graph) # (E,)
r_features = self.radial_basis(r) # (E, R)
r_features = self.envelope(r_features) # (E, R)
# 3. create neigbour embeddings
h_neighbour = index_over_neighbours(h, graph) # (E, C)
neighbour_embeddings = torch.cat(
[h_neighbour, r_features], dim=-1
) # (E, C + R)
# 4. create messages
messages = self.message(neighbour_embeddings) # (E, C)
# 5. aggregate messages
aggregated_messages = sum_over_neighbours(messages, graph) # (N, C)
# 6. update node embeddings
h = h + aggregated_messages # (N, C)
# 7. readout
local_energies = self.readout(h).squeeze(-1) # (N,)
return {"local_energies": local_energies}
Writing custom_model.py
We can now use and instantiate our model as normal:
[4]:
from ase.build import molecule
from custom_model import CustomModel
from graph_pes import AtomicGraph
model = CustomModel(cutoff=5.0, channels=3, radial_features=8)
graph = AtomicGraph.from_ase(molecule("H2O"))
model(graph)
[4]:
{'local_energies': tensor([0.6992, 0.4318, 0.4318], grad_fn=<SqueezeBackward1>)}
We get all the functionality of a GraphPESModel for free:
[5]:
model.get_all_PES_predictions(graph)
[5]:
{'energy': tensor(1.5627, grad_fn=<SumBackward1>),
'forces': tensor([[-0.0000e+00, -0.0000e+00, -1.2980e-05],
[-0.0000e+00, -7.9464e-06, 6.4898e-06],
[-0.0000e+00, 7.9464e-06, 6.4898e-06]], grad_fn=<NegBackward0>),
'local_energies': tensor([0.6992, 0.4318, 0.4318], grad_fn=<SqueezeBackward1>)}
Training¶
Let’s train our architecture on the QM7 dataset. Below, we use load-atoms to load this dataset, and save the training and validation, splits to files train.xyz and val.xyz:
[6]:
from ase.io import write
from load_atoms import load_dataset
dataset = load_dataset("QM7")
train, val, test = dataset.random_split([1000, 100, 500])
write("train.xyz", train)
write("val.xyz", val)
Our configuration file is as normal (see the graph-pes-train guide for more details):
[7]:
%%writefile config.yaml
model:
+custom_model.CustomModel:
cutoff: 5.0
channels: 3
radial_features: 8
data:
train: train.xyz
valid: val.xyz
loss: +PerAtomEnergyLoss()
fitting:
trainer_kwargs:
max_epochs: 150
accelerator: cpu
check_val_every_n_epoch: 5
optimizer:
name: AdamW
lr: 0.001
loader_kwargs:
batch_size: 64
general:
run_id: custom-model-run
progress: logged
wandb: null
Writing config.yaml
[8]:
!graph-pes-train config.yaml
[graph-pes INFO]: Started `graph-pes-train` at 2024-12-05 15:23:22.097
[graph-pes INFO]: Successfully parsed config.
[graph-pes INFO]: ID for this training run: custom-model-run
[graph-pes INFO]:
Output for this training run can be found at:
└─ graph-pes-results/custom-model-run
├─ logs/rank-0.log # find a verbose log here
├─ model.pt # the best model
├─ lammps_model.pt # the best model deployed to LAMMPS
└─ train-config.yaml # the complete config used for this run
GPU available: True (mps), used: False
TPU available: False, using: 0 TPU cores
HPU available: False, using: 0 HPUs
[graph-pes INFO]: Logging to graph-pes-results/custom-model-run/logs/rank-0.log
[graph-pes INFO]: Preparing data
[graph-pes INFO]: Setting up datasets
[graph-pes INFO]: Pre-fitting the model on 1,000 samples
[graph-pes INFO]: Number of learnable params : 159
[graph-pes INFO]: Sanity checking the model...
[graph-pes INFO]: Starting fit...
valid/metrics valid/metrics timer/its_per_s timer/its_per_s
epoch time per_atom_energy_rmse energy_rmse train valid
5 0.9 0.95875 15.93489 142.85715 225.00000
10 1.9 0.86565 12.73514 125.00000 250.00000
15 2.8 0.83548 12.34085 142.85715 225.00000
20 3.8 0.80516 11.67857 142.85715 225.00000
25 4.7 0.75636 11.28497 142.85715 250.00000
30 5.6 0.67931 9.89885 142.85715 266.66669
35 6.6 0.47443 6.82189 125.00000 225.00000
40 7.6 0.30103 4.54500 125.00000 225.00000
45 8.6 0.29590 4.52308 90.90909 183.33334
50 9.6 0.30439 4.76209 111.11111 250.00000
55 10.5 0.28471 4.38400 142.85715 225.00000
60 11.7 0.28413 4.37031 125.00000 200.00000
65 12.6 0.27077 4.18555 142.85715 266.66669
70 13.5 0.26347 4.06702 142.85715 225.00000
75 14.5 0.25786 3.99087 111.11111 225.00000
80 15.4 0.25494 3.93345 142.85715 250.00000
85 16.4 0.24410 3.77900 125.00000 225.00000
90 17.3 0.23976 3.70559 125.00000 208.33334
95 18.3 0.23695 3.71650 142.85715 225.00000
100 19.2 0.23006 3.61704 142.85715 225.00000
105 20.2 0.21487 3.33465 125.00000 225.00000
110 21.1 0.20718 3.20724 166.66667 225.00000
115 22.1 0.20142 3.11764 125.00000 225.00000
120 23.1 0.18958 2.94265 142.85715 250.00000
125 24.0 0.18257 2.83350 142.85715 291.66669
130 25.1 0.17456 2.71116 142.85715 225.00000
135 26.3 0.16515 2.58272 125.00000 225.00000
140 27.2 0.16033 2.48846 142.85715 250.00000
145 28.2 0.15938 2.47974 111.11111 208.33334
150 29.2 0.14885 2.31319 142.85715 225.00000
`Trainer.fit` stopped: `max_epochs=150` reached.
[graph-pes INFO]: Loading best weights from "graph-pes-results/custom-model-run/checkpoints/best.ckpt"
[graph-pes INFO]: Training complete. Awaiting final Lightning and W&B shutdown...
Analysis¶
We can now load our trained model and analyse the results:
[9]:
from graph_pes.models import load_model
model = load_model("graph-pes-results/custom-model-run/model.pt")
model
[9]:
CustomModel(
(Z_embedding): PerElementEmbedding(dim=3, elements=[])
(radial_basis): Bessel(n_features=8, cutoff=5.0, trainable=True)
(envelope): PolynomialEnvelope(cutoff=5.0, p=6)
(message): MLP(11 → 6 → 6 → 3, activation=CELU(alpha=1.0))
(readout): MLP(3 → 3 → 1, activation=CELU(alpha=1.0))
)
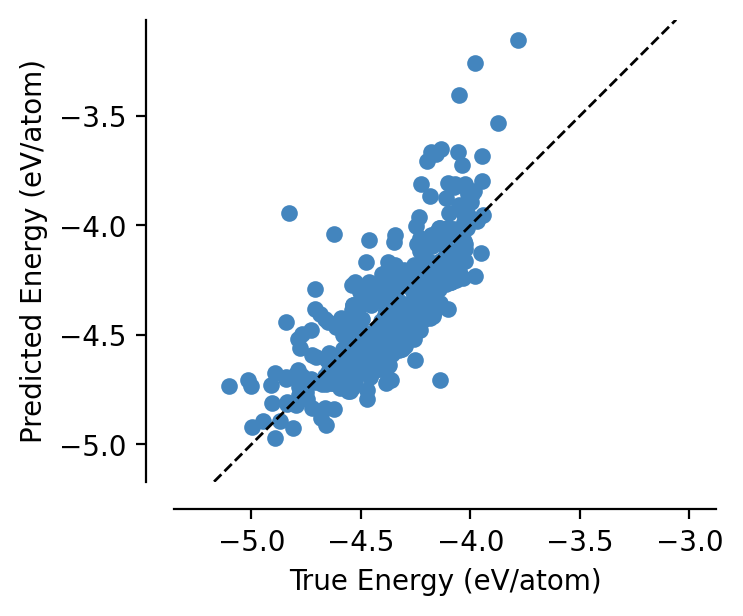
This is a very unsophisticated architecture, so we don’t expect it to be particularly accurate…
[10]:
from graph_pes.utils.analysis import parity_plot
%config InlineBackend.figure_format = 'retina'
parity_plot(
model,
test,
property="energy_per_atom",
units="eV/atom",
)
MD¶
Running MD with our new model architecture is straightforward.
Below, we use ASE-driven MD for simplicity - please see the LAMMPS MD guide for instructions on how to run MD with our model in LAMMPS.
[18]:
from ase import units
from ase.build import molecule
from ase.md.langevin import Langevin
# set up calculator and structure
calculator = model.ase_calculator()
structure = molecule("CH4")
structure.center(vacuum=3.0) # place in a large unit cell
structure.pbc = True
# set up MD
structure.calc = calculator
dynamics = Langevin(
structure,
timestep=0.1 * units.fs,
temperature_K=300,
friction=0.01 / units.fs,
)
dynamics.attach(
lambda: structure.write("dump.xyz", append=True),
interval=100,
)
# run MD
dynamics.run(1000) # 1000 steps = 0.1 ps
[18]:
True
Unsurprisingly, our very simple model has not been able to keep the simulation stable:
[19]:
from load_atoms import view
view(structure, show_bonds=True)
[19]: