🙋♂️ First Author Works:
Synthetic pre-training for neural-network interatomic potentials
J. Gardner, K. Baker and V. Deringer
Machine Learning: Science and Technology 2024

Machine learning (ML) based interatomic potentials have transformed the field of atomistic materials modelling. However, ML potentials depend critically on the quality and quantity of quantum-mechanical reference data with which they are trained, and therefore develop- ing datasets and training pipelines is becoming an increasingly central challenge. Leveraging the idea of “synthetic” (artificial) data that is common in other areas of ML research, we here show that synthetic atomistic data, themselves obtained at scale with an existing ML po- tential, constitute a useful pre-training task for neural-network interatomic potential models. Once pre-trained with a large synthetic dataset, these models can be fine-tuned on a much smaller, quantum-mechanical one, improving numerical accuracy and stability in computa- tional practice. We demonstrate feasibility for a series of equivariant graph-neural-network potentials for carbon, and we carry out initial experiments to test the limits of the approach.
Synthetic data enable experiments in atomistic machine learning
J. Gardner, Z. Faure Beaulieu and V. Deringer
Journal of Chemical Physics 2023
🔖Paper — 📝Pre-print — 🤖Code — 💿Data

Machine-learning models are increasingly used to predict properties of atoms in chemical systems. There have been major advances in developing descriptors and regression frameworks for this task, typically starting from (relatively) small sets of quantum-mechanical reference data. Larger datasets of this kind are becoming available, but remain expensive to generate. Here we demonstrate the use of a large dataset that we have “synthetically” labelled with per-atom energies from an existing ML potential model. The cheapness of this process, compared to the quantum-mechanical ground truth, allows us to generate millions of datapoints, in turn enabling rapid experimentation with atomistic ML models from the small- to the large-data regime. This approach allows us here to compare regression frameworks in depth, and to explore visualisation based on learned representations. We also show that learning synthetic data labels can be a useful pre-training task for subsequent fine-tuning on small datasets. In the future, we expect that our open-sourced dataset, and similar ones, will be useful in rapidly exploring deep-learning models in the limit of abundant chemical data.
📖 Other Publications:
An automated framework for exploring and learning potential-energy surfaces
Y. Liu, J. Morrow, C. Ertural, N. Fragapane, J. Gardner, A. Naik, Y. Zhou, J. George, V Deringer
arXiv 2024 (in peer review)
Machine learning has become ubiquitous in materials modelling and now routinely enables large-scale atomistic simulations with quantum-mechanical accuracy. However, developing machine-learned interatomic potentials requires high-quality training data, and the manual generation and curation of such data can be a major bottleneck. Here, we introduce an automated framework for the exploration and fitting of potential-energy surfaces, implemented in an openly available software package that we call autoplex (`automatic potential-landscape explorer’). We discuss design choices, particularly the interoperability with existing software architectures, and the ability for the end user to easily use the computational workflows provided. We show wide-ranging capability demonstrations: for the titanium-oxygen system, SiO2, crystalline and liquid water, as well as phase-change memory materials. More generally, our study illustrates how automation can speed up atomistic machine learning – with a long-term vision of making it a genuine mainstream tool in physics, chemistry, and materials science.
Data as the next challenge in atomistic machine learning
C. Ben Mahmoud, J. Gardner and V. Deringer
Nature Computational Science 2024
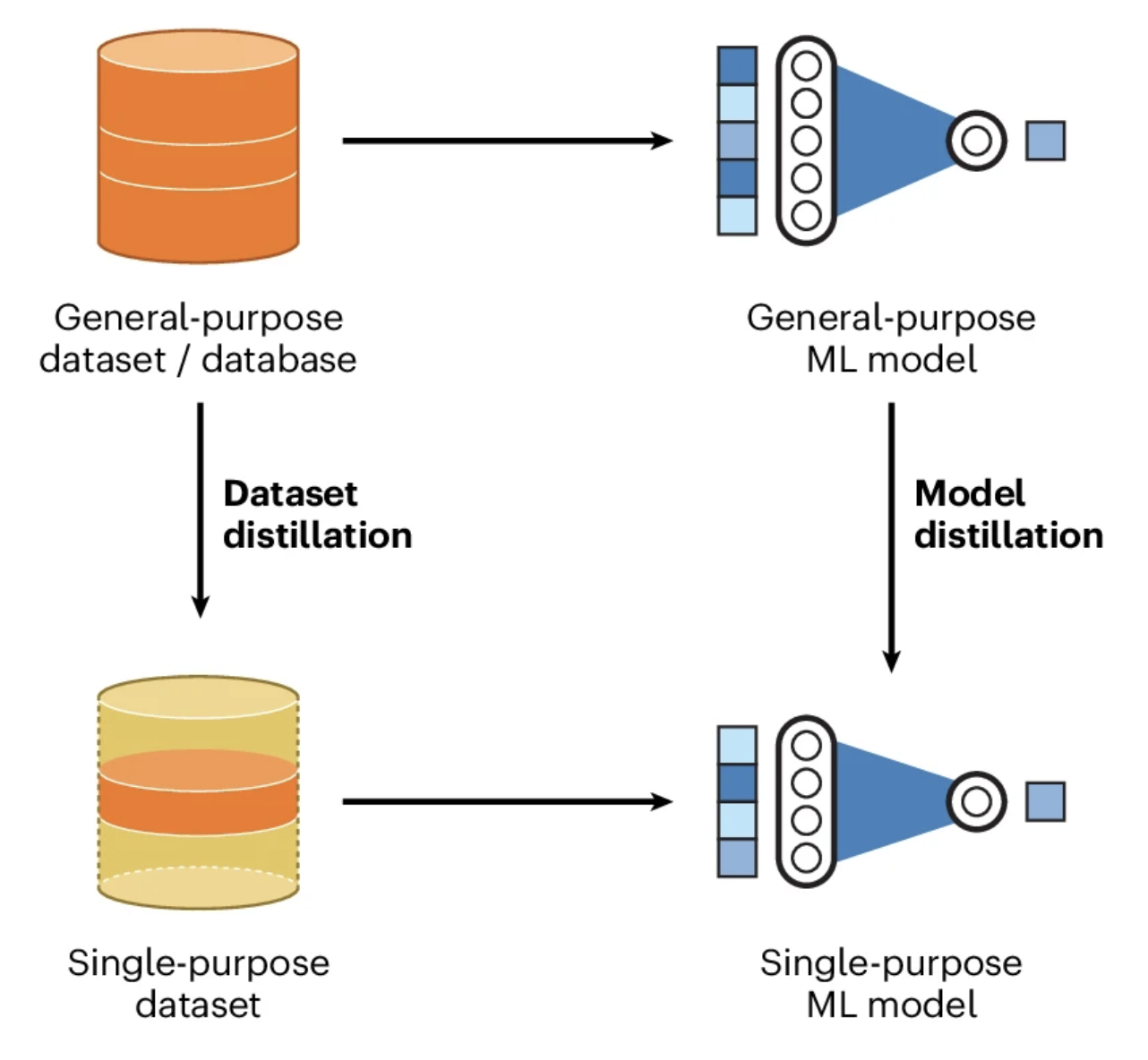
As machine learning models are becoming mainstream tools for molecular and materials research, there is an urgent need to improve the nature, quality, and accessibility of atomistic data. In turn, there are opportunities for a new generation of generally applicable datasets and distillable models.
Coarse-grained versus fully atomistic machine learning for zeolitic imidazolate frameworks
Z. Faure Beaulieu, T. Nicholas, J. Gardner, A. Goodwin and V. Deringer
Chemical Communications 2023
🔖Paper — 📝Pre-print — 🤖Code — 💿Data
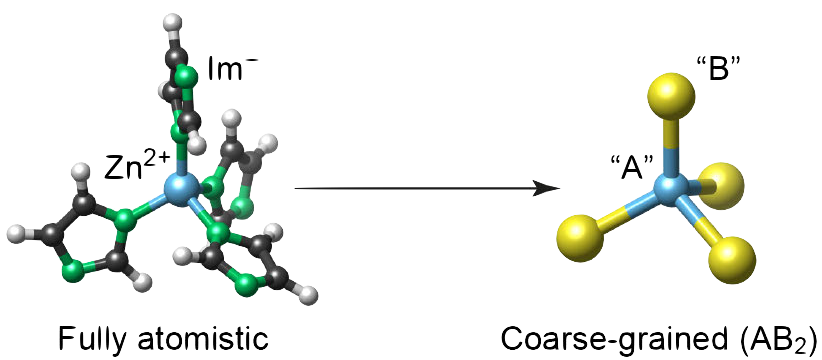
Zeolitic imidazolate frameworks are widely thought of as being analogous to inorganic AB₂ phases. We test the validity of this assumption by comparing simplified and fully atomistic machine-learning models for local environments in ZIFs. Our work addresses the central question to what extent chemical information can be “coarse-grained” in hybrid framework materials.
How to validate machine-learned interatomic potentials
J. Morrow, J. Gardner and V. Deringer
Journal of Chemical Physics 2023
Machine learning (ML) approaches enable large-scale atomistic simulations with near-quantum-mechanical accuracy. With the growing availability of these methods there arises a need for careful validation, particularly for physically agnostic models - that is, for potentials which extract the nature of atomic interactions from reference data. Here, we review the basic principles behind ML potentials and their validation for atomic-scale materials modeling. We discuss best practice in defining error metrics based on numerical performance as well as physically guided validation. We give specific recommendations that we hope will be useful for the wider community, including those researchers who intend to use ML potentials for materials “off the shelf”.
Using spectroscopy to probe relaxation, decoherence, and localization of photoexcited states in π-conjugated polymers
W. Barford, J. Gardner, and J. R. Mannouch
Faraday Discussions 2020

We use the coarse-grained Frenkel-Holstein model to simulate the relaxation, decoherence, and localization of photoexcited states in conformationally disordered π-conjugated polymers. The dynamics are computed via wave-packet propagation using matrix product states and the time evolution block decimation method. The ultrafast (i.e., t < 10 fs) coupling of an exciton to C–C bond vibrations creates an exciton–polaron. The relatively short (ca. 10 monomers) exciton-phonon correlation length causes ultrafast exciton-site decoherence, which is observable on conformationally disordered chains as fluorescence depolarization. Dissipative coupling to the environment (modelled via quantum jumps) causes the localization of quasi-extended exciton states (QEESs) onto local exciton ground states (LEGSs, i.e., chromophores). This is observable as lifetime broadening of the 0–0 transition (and vibronic satellites) of the QEES in two-dimensional electronic coherence spectroscopy. However, as this process is incoherent, neither population increases of the LEGSs nor coherences with LEGSs are observable.
🎤 Talks and Posters
- Talk and slides presented at the LJC MLIP workshop, Cambridge, 2025
- Poster presented at the 2023 NeurIPs @ Cambridge meetup
- Talk and slides presented at the 2023 SIMPLAIX Workshop for “Machine Learning for Multiscale Molecular Modeling”.
- Poster presented at the 2023 CECAM PsiK Research Conference.